- Wnt-Frizzled signalling in synaptic assembly during development
- Wnt signalling in structural and functional synaptic plasticity in the CNS
- Dysfunction of Wnt signalling in Alzheimer’s disease and its contribution to synapse degeneration
- How do synapses become resilient to toxic molecules in the adult and aging brain?
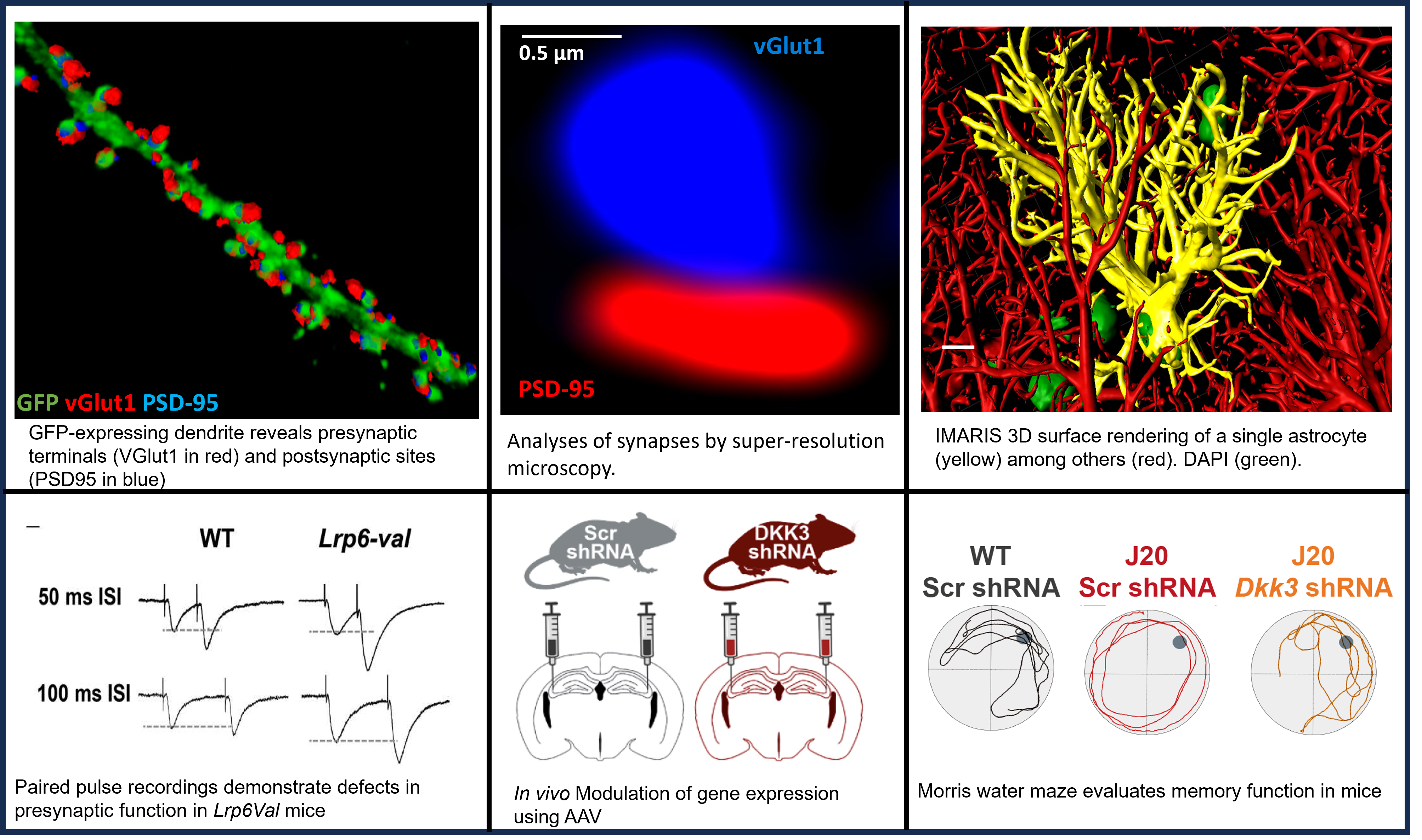
The overarching aim of our research program is to elucidate the cellular and molecular mechanisms that regulate the formation, function and maintenance of synaptic connections in the mammalian nervous system. We are particularly interested in the role of Wnt signalling in these processes.
We and others have demonstrated that this prominent pathway promotes the formation of excitatory synapses during postnatal development and is required for long-term potentiation in the adult hippocampus. We also found that the Wnt cascade is crucial for synapse integrity in the adult brain as deficits in Wnt signalling cause synaptic plasticity defects, synapse degeneration and defects in memory. Notably, we discovered that Wnt signalling is compromised in Alzheimer’s disease (AD), contributing to synapse degeneration and memory impairment.
Understanding the molecular mechanisms that control the formation and maintenance of synapses during development and in the adult and aging brain is crucial for developing strategies for the treatment of neurodevelopmental disorders and neurodegenerative diseases where synapses are affected.