Increasing evidence suggests that canonical Wnt signalling is compromised in AD. The first evidence linking deficiency in Wnt signalling and AD came from the finding that Dickkopf-1 (Dkk1), an endogenous and specific secreted Wnt antagonist, is increased in the human AD brain and in the brain of AD mouse models. Interestingly, Dkk3, another member of the Dickkopf Wnt antagonist family, is also elevated in the brains of AD patients and it is accumulated in Aβ plaques. Another evidence for a link between Wnt signalling and AD comes from the discovery of a genetic link between variants of the Wnt co-receptor LRP6 and late-onset AD (LOAD). Moreover, our recent findings indicate that several Wnt receptors from the Frizzled family are downregulated in human AD brains due to the epigenetic repression of these genes. Altogether, the evidence points to Wnt signalling as a potential target in AD.
Wnt antagonists and AD
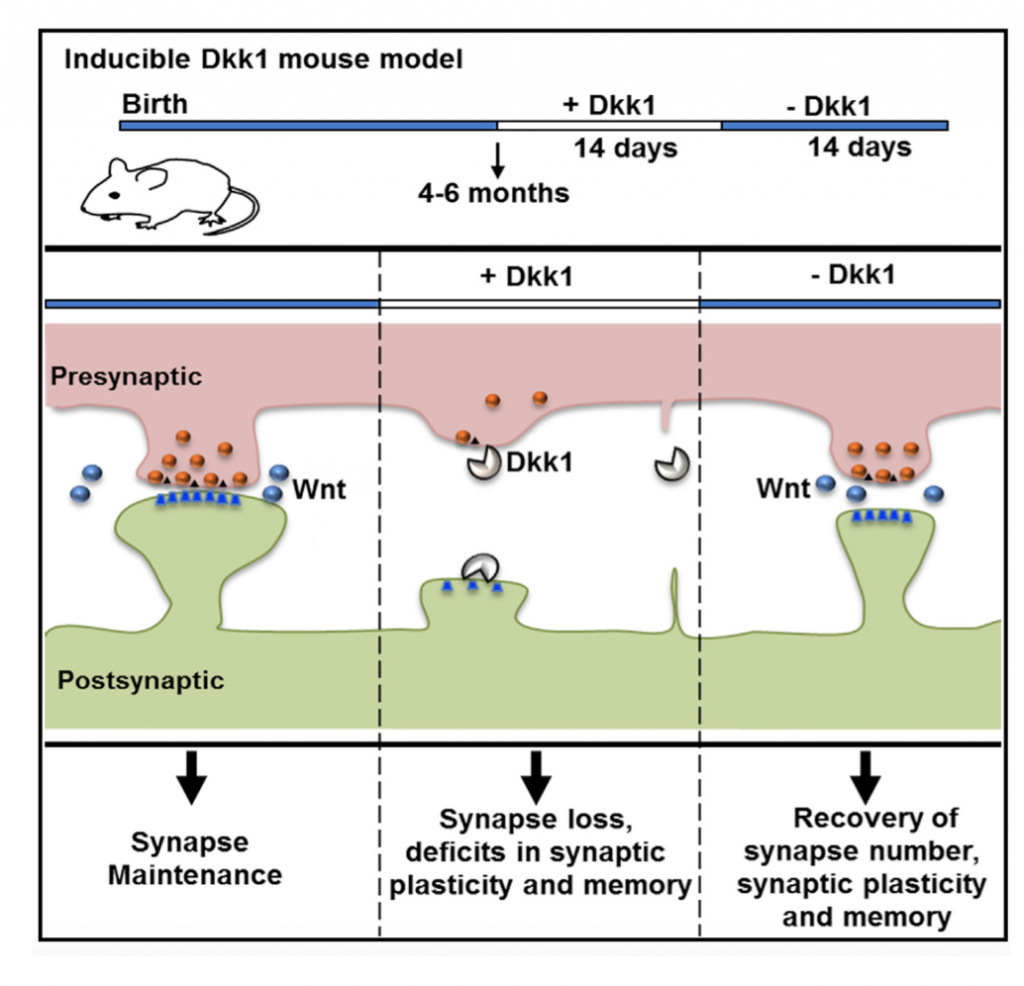
Our studies showed that Amyloid-β oligomers (Aβo) upregulate the expression of Dkk1. Importantly, blockade or knockdown of Dkk1 protects synapses from Aβ toxicity (Purro et al, J.Neuroscience 2012). In vivo, induction of Dkk1 for two weeks in the adult brain leads to a significant loss of excitatory synapses, blockade of long-term potentiation (LTP), enhanced long-term depression (LTD) and long-term memory defects. Importantly, reactivation of the Wnt pathway, after substantial synapse degeneration, results in the full recovery of the phenotype (Marzo et al, 2016). Thus, we have demonstrated that endogenous Wnt signalling is required for the integrity of synapses and memory circuits in the adult hippocampus. Our research suggests that modulation of the Wnt pathway could provide an effective approach for restoring neuronal connectivity at early stages of AD.
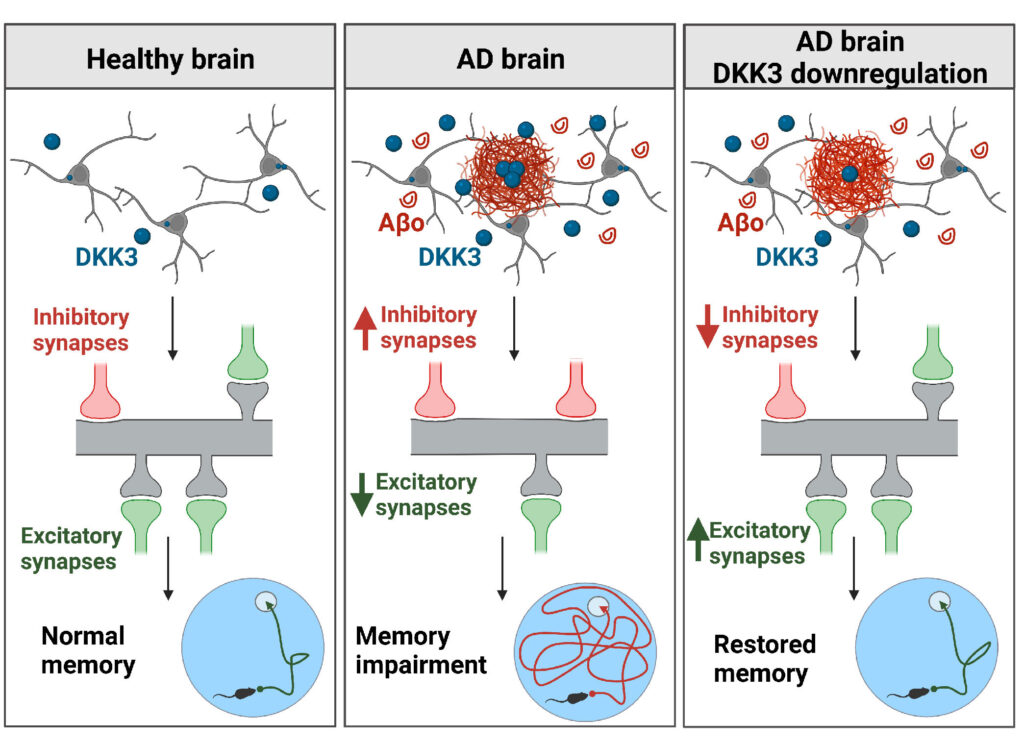
We are also interested in Dkk3, another member of the Dkk protein family of Wnt antagonists. This secreted protein is present at amyloid-β plaques in the human brain. Moreover, recent studies showed that Dkk3 levels are increased in the plasma and CSF of AD patients and that its level is increased in several brain-affected areas and cortical synaptosomes from AD subjects. We evaluated the contribution of Dkk3 in AD using human brain samples, mouse models of AD and in vivo genetic approaches to determine the role of this Wnt antagonist. We found that DKK3 expression is upregulated in the brains of AD subjects and that DKK3 protein levels increase at early stages in human AD. Importantly, Dkk3 results in the degeneration of excitatory synapses and an increase in inhibitory ones, thus resulting in synaptic imbalance. In contrast, knockdown of Dkk3 in vivo using AAV vectors results in the restoration of both excitatory and inhibitory synapse number and memory in a mouse model of AD (Martin-Flores et al, eLife 2024). These exciting results demonstrate that increased activation of Wnt signalling is a viable approach to restore neuronal connectivity in AD.
Wnt receptors and AD
As mentioned before, there is a genetic link between variants of the LRP6 co-receptor and LOAD. One of the variants, called LRP6Val-1062, carries a substitution from isoleucine to valine, conferring decreased Wnt signalling in cell lines. We used CRISPR-Cas9 genome editing to generate a novel knock-in mouse model to examine the impact of a mutation in LRP6 on synapse degeneration and AD pathogenesis. Mice carrying the LRP6-val mutant first exhibit defects in synapse function followed by a later degeneration of excitatory synapses in the hippocampus in ageing. Importantly, we found that the presence of this LRP6 variant in an AD mouse model such as NLGF results in increased synapse degeneration (Jones et al, Science Advances 2023). Thus, LRP6-val increases the risk of synapse degeneration in AD.
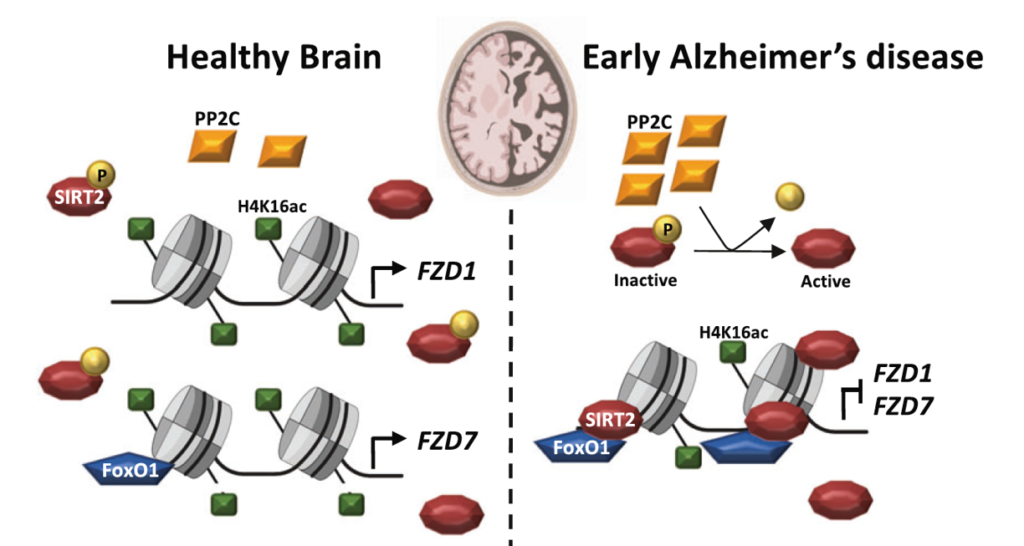
Our recent studies have shown that several Wnt components are deregulated in AD at the mRNA level. These findings led us to investigate if epigenetic mechanisms could contribute to this deregulation. Indeed, using human brain samples and established AD mouse models we showed that several Wnt receptors are deregulated through epigenetic mechanisms. We investigated possible epigenetic changes using chromatin immuno-precipitation (ChIP) analyses in AD mouse models and in the human AD brain combined with genetic and pharmacological approaches. Our results showed that Wnt receptors are epigenetically repressed in AD. Specifically, there is Sirtuin2-induced H4K16ac deacetylation of Frizzled1 and Frizzled7 promoters, resulting in the downregulation of these synaptic receptors in AD (Palomer et al, Mol Psychiatry 2022).